
Eine neue route zu blockieren Kinder-Knochen-Krebs
Das Ewing-Sarkom ist ein Knochenkrebs, erscheint vor allem bei Jugendlichen auf. Durch ein defektes gen, sobald es breitet sich auf entfernte Organe es ist schwer zu behandeln. Forscher am Weizmann Institute of Science haben entdeckt, molekulare Interaktionen zugrunde liegenden Ewing-Sarkome und vorgeschlagen, eine mögliche Behandlung, die gezeigt hat Versprechen in einer Studie an Mäusen. Diese Ergebnisse wurden vor kurzem veröffentlicht in Cell Reports.
Postdoc-Stipendiat Dr. Swati Srivastava im Labor von Prof. Yosef Yarden, der in die Biologische Regulation-Abteilung zusammen mit Kollegen durchgeführt, die Forschung konzentriert sich auf die Rezeptoren für steroid-Hormone genannt Glukokortikoide. Diese Rezeptoren sind in fast allen menschlichen Zellen, Vermittlung von hormonellen Botschaften mit stress, Wachheit und eine Vielzahl von anderen wichtigen Funktionen. Aber manchmal Glukokortikoid-Rezeptoren stimulieren Maligne Wachstum. Sie tun dies, indem auf den Zellkern, wo Sie physisch interagieren und die Bindung mit Transkriptionsfaktoren—Moleküle schalten Gene an oder aus. Die Forscher wollten mehr über die Rolle dieser Interaktion in Malignität.
Eine hoch empfindliche protein-Interaktions-Analyse geeignet für lebende Zellen ergab, die zuvor unbekannte Wechselwirkungen: nach der Aktivierung von Hormonen, Glukokortikoid-Rezeptoren gefunden wurden, um die Bindung im Zellkern an Transkriptionsfaktoren der E-twenty-six, oder die ETS-Familie, die zusammen einen physikalischen Komplex. Einer der Transkriptionsfaktoren der ETS-Familie ist bekannt, um die Entwicklung voranzutreiben von Ewing-Sarkom; Ihre gen-sicherungen ungewöhnlich mit einem anderen gen, erstellen ein Onkogen: ein krebserregendes gen.
Bei der Studie stellte sich diese Verbindung zwischen der Ewing-Sarkom-Onkogen und Glukokortikoid-Rezeptoren, die Forscher getestet die Hypothese:, dass diese Rezeptoren regt das Wachstum von Ewing-Sarkom. Eine Reihe von Studien Beweise geliefert, dass dies tatsächlich der Fall ist. Physikalische Bindung zwischen den Glukokortikoid-Rezeptoren und des proteins, das durch das Onkogen erhöht das Wachstum und die migration von Ewing-Sarkom-Zellen im Labor Gericht und gab einen noch stärkeren Schub für das Wachstum und die Ausbreitung des Sarkom in Labor-Mäuse.
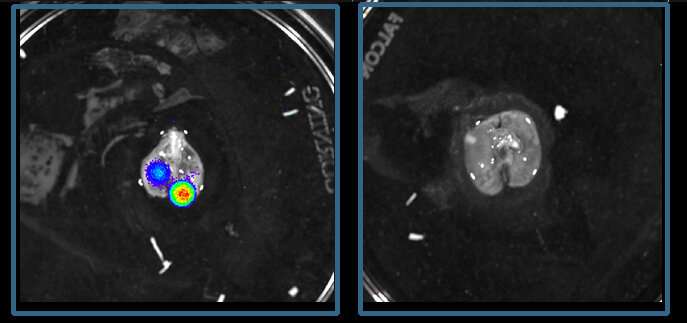
Die große medizinische Bedeutung dieser Ergebnisse ist, dass Sie öffnen die Tür zu einer neuen Therapie-option für das Ewing-Sarkom. Wenn die Forscher implantierten humanen Ewing-Sarkom-Zellen in Mäusen, die Tumoren wuchsen sehr viel langsamer als die Mäuse wurden behandelt mit metyrapone, ein Medikament, das zugelassen ist für die Behandlung von Nebennieren-Insuffizienz und funktioniert durch die Verringerung der Synthese von glucocorticoiden. In anderen Experimenten, auch in Mäusen, ein weiteres Medikament, mifepristone, die Blöcke des Glukokortikoid-rezeptor und wird genehmigt für andere klinische Anwendungen, verhindert die Metastasierung des Ewing-Sarkom über eine große cancer cell dissemination route von Knochen in die Lunge. Im Gegensatz, wenn die Forscher erhöhte sich die Aktivität des Glukokortikoid-Rezeptoren, die Sarkome wuchs und breitete sich viel schneller.
Darüber hinaus die Forscher führten eine genetische Analyse von Tumorproben von Patienten mit Ewing-Sarkom und ermittelten sieben Gene durch die Glukokortikoid-Rezeptoren, exprimiert in höher-als-normale Niveaus bei Patienten mit besonders tödlichen Tumoren. Diese Gene dienen als genetische Signatur ermöglicht eine Auswahl von Patienten für die Behandlung: wer hochreguliert „Signatur“ die Gene sind besonders wahrscheinlich, profitieren von der Behandlung zur Neutralisierung der Glukokortikoid-Rezeptoren. Die Signatur-Gene kann auch helfen, vorherzusagen, den Verlauf der Erkrankung: Ihre erhöhte expression kann das signal einer schlechten Prognose; verringerte expression, auf der anderen Seite, kann das signal besser die überlebenschancen.
Wenn die Forschung an menschlichen Patienten bestätigt die Ergebnisse der Studie, können Sie bieten eine neue Hoffnung für Kinder und Jugendlichen mit malignen Erkrankungen, besonders in Fällen, wenn das Sarkom hat sich Metastasen über den Knochen hinaus.